Trusted By
120,000+ People

El mieloma múltiple es la segunda neoplasia hematológica en orden de frecuencia. Se define por la presencia de células plasmáticas monoclonales con capacidad para producir una paraproteína monoclonal y causar alteraciones clínicas en forma de anemia, insuficiencia renal, hipercalcemia o lesiones óseas.
El mieloma múltiple (MM), aunque es una enfermedad rara, es la segunda neoplasia hematológica más común. Se encuentra en el espectro de discrasias de células plasmáticas, que comienza con una gammapatía monoclonal de importancia desconocida (MGUS) para revelar leucemia de células plasmáticas y mieloma extramedular. El MM está asociado con una morbilidad significativa debido a la destrucción del órgano final. Es una enfermedad de la población de edad avanzada y su incidencia en la población afroamericana es el doble que la de la población europea americana.
La edad media al momento del diagnóstico es de 65 años. La presentación en menores de 40 años ocurre en menos del 3% de los pacientes.
Aunque la etiología de MM sigue siendo esquiva, las características clínicas, los patrones de incidencia de disparidad racial observados, la agrupación familiar informada y la incidencia más joven en AA sugieren un papel para los genes de susceptibilidad. Las diferencias raciales descritas no pueden atribuirse únicamente a las diferencias socioeconómicas y de acceso a la atención y eso, al menos en parte, probablemente se deba a diferencias genéticas y biológicas subyacentes más profundas. Para comprender mejor esto, Baker et al. intentaron asociar alteraciones genómicas específicas a las diferencias clínicas raciales observadas. Los autores utilizaron tres plataformas para evaluar diferentes marcadores pronósticos: hibridación fluorescente in situ (FISH), hibridación genómica comparativa basada en matriz (aCGH) y perfil de expresión génica (GEP). La única diferencia significativa que observaron fue una menor frecuencia de translocaciones de IgH. Sin embargo, el estudio fue confundido por un pequeño número de AA analizados (11 casos para FISH).
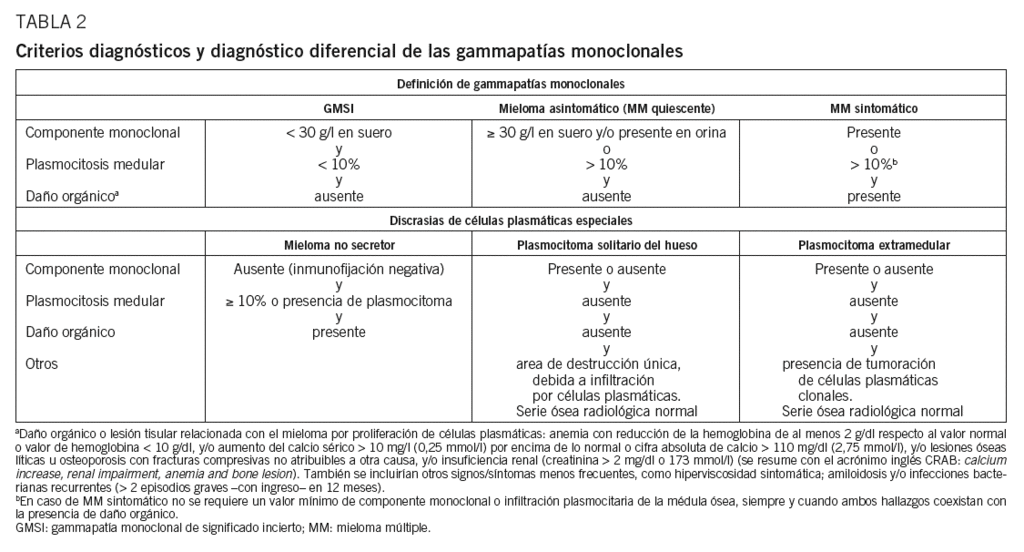
El desarrollo de MM suele estar precedido por una gammapatía monoclonal de significado incierto (MGUS por sus siglas en ingles), una condición premaligna que se produce cuando las células plasmáticas sufren mutaciones que restauran su capacidad de proliferación. En la MGUS, estas células plasmáticas clonales ocupan menos del 10% de la médula ósea. El valor de la proteína sérica es inferior a 3 g / dL y el daño al órgano terminal relacionado con el mieloma está ausente. Una etapa de enfermedad intermedia entre MGUS y MM, denominada MM latente, se caracteriza por un nivel de proteína M de 3 g / dL o más y más del 10% de células plasmáticas clonales en la médula ósea, pero sin síntomas de daño en el órgano terminal relacionado con el mieloma.
Una variedad de anormalidades citogenéticas se encuentran en la MGUS y el MM. Aproximadamente la mitad de los casos son hiperdiploides, generalmente con copias adicionales de los cromosomas impares. La mayoría del resto son no hiperdiploides y se caracterizan por una translocación primaria que involucra el gen de la cadena pesada de Ig en 14q32. Además, prácticamente todos los casos involucran desregulación de la vía ciclina D / retinoblastoma (ciclina D / RB). Esta heterogeneidad genética contribuye a la rápida aparición de resistencia a los medicamentos en MM.
La evidencia creciente sugiere que el microambiente de la médula ósea de las células tumorales desempeña un papel fundamental en la patogénesis de los mielomas. Este descubrimiento ha resultado en la expansión de las opciones de tratamiento. El papel de las citocinas en la patogénesis de MM es un área importante de investigación. La interleucina (IL) -6 también es un factor importante que promueve el crecimiento in vitro de las células de mieloma. Otras citocinas son el factor de necrosis tumoral y la IL-1b.
La base fisiopatológica de las secuelas clínicas de MM implica los sistemas esquelético, hematológico, renal y nervioso, así como los procesos generales.
El dolor óseo es el síntoma más típico y frecuente. Aparece en el 60-80% de los pacientes en el momento del diagnóstico, generalmente en la columna vertebral, pero también en el esternón, las costillas o la zona proximal de las extremidades. Su origen son lesiones óseas que, por radiología, suelen corresponder a típicas imágenes osteolíticas en «sacabocados» sin esclerosis periférica, pero también pueden manifestarse como osteoporosis grave o fracturas patológicas. Las osteólisis son incluso más frecuentes que el dolor, pues aparecen en el 80% de los casos y en el resto hay lesiones microscópicas que, aun siendo invisibles en la radiografía convencional, pueden dar imágenes visibles por resonancia magnética (RM). Se localizan sobre todo en huesos con gran cantidad de médula ósea, es decir, por orden de frecuencia: cráneo, columna vertebral, costillas, pelvis y epífisis proximales de huesos largos. El origen de estas lesiones hay que buscarlo en un desequilibrio entre resorción y formación óseas. En etapas iniciales se observa un aumento de la resorción por activación de osteoclastos debida al aumento de estimulantes como el factor de necrosis tumoral alfa y beta, la interleucina (IL) 1-ß e IL-6, pero sobre todo a una hiperactividad del receptor del activador del factor nuclear kappa y su ligando (RANK/RANK-L), ya que el ligando se expresa en células plasmáticas y estroma. El aumento destructivo se compensa al principio con un aumento de la formación ósea, pero al crecer el clon tumoral se sintetiza Dickkopf-1 (DKK1), que es un factor que inhibe la vía de Wingless tipo-1 (WNT1), esencial en los osteoblastos. Así pues, la formación ósea se resiente y se acaba por generar un desequilibrio que ocasiona una pérdida neta de masa ósea, lo que genera la clásica tríada de la lesión ósea del mieloma: dolor, hipercalcemia y fracturas patológicas. La hipercalcemia está presente en un tercio de los pacientes en el momento del diagnóstico y un 30% adicional la desarrolla durante la evolución de la enfermedad. Las fracturas patológicas más frecuentes aparecen en la columna vertebral y pueden motivar lesiones radiculares y/o medulares, aunque en estos casos suele haber un plasmocitoma asociado.
Otra manifestación clínica clásica del mieloma es la anemia. Hasta el 50% de los pacientes presenta anemia moderada (aproximadamente 10 g/dl) y un 25% de los casos, anemia grave (< 8 g/dl)4. Como en tantas enfermedades neoplásicas, su causa es multifactorial, si bien los mecanismos más importantes son 2:
a) reducción de la concentración de eritropoyetina circulante, debido al aumento de citocinas como el factor de necrosis tumoral alfa e IL-1, y a la posible existencia de lesión renal asociada
b) presencia del ligando de Fas en la superficie de las células plasmáticas, que, al activar a Fas en los progenitores eritroides, activa la apoptosis5. Otras causas de anemia pueden ser la invasión masiva de la médula ósea, la quimioterapia, la presencia de déficit asociados de vitamina B12 y/o folato, y el efecto diluyente de la hiperproteinemia plasmática. Junto a la anemia, también son frecuentes los síntomas constitucionales (astenia y pérdida de peso).
La insuficiencia renal (IR) aparece en un 25-30% de los pacientes, aunque las series más recientes describen una frecuencia menor. Su origen es multifactorial, si bien la causa más descrita es la eliminación renal de cadenas ligeras de Ig (proteinuria de Bence-Jones), cuya presencia en los túbulos se denomina histológicamente «riñón de mieloma». Otras causas son hipercalcemia, hiperuricemia, deshidratación, infecciones urinarias de repetición, hiperviscosidad, amiloidosis e infiltración tumoral. Suele ser una IR crónica, pero también puede manifestarse como IR aguda (más raramente como síndrome de Fanconi adquirido o síndrome nefrótico).
Las infecciones son la primera causa de mortalidad del MM, con una incidencia de 0,8-2,2 episodios por paciente y año, entre 7 y 15 veces por encima de la observada en pacientes hospitalizados por otros motivos. Suelen ser bacterianas y localizadas en el pulmón y las vías urinarias, pero también las hay virales (sobre todo, herpes zóster) y fúngicas. La causa principal de la susceptibilidad infecciosa es la hipogammaglobulinemia policlonal, pero también hay otros motivos: reducción de células T CD4+, alteración funcional de las células citolíticas, anomalías en el sistema del complemento y granulopenia ocasional.
El síndrome de hiperviscosidad es raro (4-8%) en el MM. Su génesis se relaciona con la concentración de la paraproteína y con las características moleculares de ésta (es más frecuente con IgM excepcional en el MM, IgG3 e IgA). Hasta el 10-15% de los mielomas presentan amiloidosis, aunque con poca participación orgánica. Otras manifestaciones clínicas son diátesis hemorrágica, hepatomegalia (un 20% de los casos), esplenomegalia (< 10%) y plasmocitomas (tumoraciones de células plamáticas) en cualquier localización.
El diagnóstico de MM requiere la presencia de uno o más eventos definitorios de mieloma (MDE) además de la evidencia de 10% o más de células plasmáticas clonales en el examen de médula ósea o un plasmacitoma probado por biopsia. El MDE consta de características de CRAB establecidas (hipercalcemia, insuficiencia renal, anemia o lesiones óseas líticas) y tres biomarcadores específicos: células plasmáticas de la médula ósea clonales de 60% o más, relación de cadena ligera libre de suero (FLC) de 100 o más (siempre involucrado El nivel de FLC es ?100 mg / L) y más de una lesión focal en la resonancia magnética (MRI). Cada uno de los nuevos biomarcadores está asociado con un riesgo de progresión de aproximadamente 80% a daño sintomático de los órganos terminales en dos o más estudios independientes.
Tras una sospecha clínica o como hallazgo casual, la investigación de un paciente con MM requiere una serie de exploraciones complementarias obligadas para confirmar el diagnóstico, establecer el pronóstico y orientar el tratamiento. En la tabla 1 se exponen las exploraciones necesarias según el momento diagnóstico concreto del paciente
Aunque el MM todavía se considera una enfermedad única, en realidad es una colección de varias neoplasias malignas de células plasmáticas citogenéticamente distintas. Las trisomías y las translocaciones de IgH se consideran anormalidades citogenéticas primarias y ocurren en el momento del establecimiento de la gammapatía monoclonal de significado incierto. Otros cambios citogenéticos denominados anormalidades citogenéticas secundarias ocurren durante el curso de la enfermedad, como ganancia (1q), del (1p), del (17p), del (13), mutaciones RAS y translocaciones secundarias que involucran MYC. Se considera que la presencia de del (17p), ganancia (1q), t (4; 14), t (14; 16) yt (14; 20) refleja una enfermedad de alto riesgo.
La supervivencia al MM ha mejorado significativamente en los últimos 15 años. Existen muchos medicamentos activos para tratar el MM además de los alquilantes y los corticosteroides. La talidomida, la lenalidomida y la pomalidomida se denominan agentes inmunomoduladores (IMiD). Bortezomib, carfilzomib e ixazomib son inhibidores del proteasoma. Elotuzumab y daratumumab son anticuerpos monoclonales dirigidos a SLAMF7 y CD38, respectivamente. Panobinostat es un inhibidor de la desacetilasa.
Se han desarrollado numerosos regímenes con estos nuevos medicamentos, y cada año, se están desarrollando nuevos regímenes adicionales. Los datos recientes muestran que el estado negativo de la enfermedad residual mínima (MRD por sus siglas en inglé), según lo estimado por los métodos moleculares de próxima generación o la citometría de flujo, tiene un valor pronóstico favorable. Sin embargo, se necesitan ensayos adicionales para determinar si es necesario realizar cambios en el tratamiento base del estado de MRD. En la actualidad, los resultados de MRD se recomiendan principalmente como una medida de pronóstico y no para su uso en la toma de decisiones de tratamiento.
La situación del tratamiento en países no desarrollados
Aunque las mejores opciones terapéuticas para el tratamiento de personas con MM se han demostrado en ensayos aleatorios, los nuevos medicamentos son cada vez más costosos y muchos agentes y combinaciones no están ampliamente disponibles en países con circunstancias desfavorecidas. Por ejemplo, mientras que la talidomida está ampliamente disponible a un bajo costo, la lenalidomida es más costosa y la pomalidomida es muy costosa; Mientras que el bortezomib es moderadamente caro, el ixazomib y el carfilzomib son muy costosos. Los agentes más nuevos, como daratumumab, elotuzumab, panobinostat y selinexor, son extremadamente caros y, por lo general, no están disponibles.
En un estudio realizado por el equipo del Dr. Guillermo Ruiz Argüelles, de determinó que a pesar del uso de medicamentos que no eran especialmente caros, se observaron buenos resultados en pacientes con MM recién diagnosticado, con una SG de más de 13 años (mediana aún no alcanzada). Al analizar la Supervivencia Global de los pacientes incluidos en este estudio y separados por intervalos de 5 años, la supervivencia continuó mejorando desde 1993, lo que sugiere que las medidas generales de apoyo, que también han mejorado con el tiempo, también pueden haber contribuido al mejor resultado de los pacientes.
El MM es una enfermedad heterogénea, con una supervivencia que varía de 1 año a más de 10 años. La mediana de supervivencia en pacientes no seleccionados con MM es de 3 años. La tasa de supervivencia relativa a 5 años es del 46,6%. La supervivencia es mayor en las personas más jóvenes y menor en los ancianos.
La carga tumoral y la tasa de proliferación son los dos indicadores clave para el pronóstico en pacientes con MM. Se han publicado muchos esquemas para ayudar a determinar el pronóstico. Un esquema utiliza la proteína C reactiva (PCR) y la microglobulina beta-2 (que es una expresión de la carga tumoral) para predecir la supervivencia:
Los factores pronósticos pobres incluyen los siguientes:
El pronóstico de acuerdo al tratamiento es el siguiente:
Las infecciones son una causa importante de muerte prematura en MM. En un estudio del Reino Unido, el 10% de los pacientes fallecieron dentro de los 60 días posteriores al diagnóstico de MM, y el 45% de esas muertes se debieron a una infección. En un estudio sueco, el 22% de los pacientes fallecieron por infección dentro del primer año después del diagnóstico. El riesgo de infecciones bacterianas (p. Ej., Meningitis, septicemia, neumonía) e infecciones virales (p. Ej., Herpes zoster, influenza) fue siete veces mayor en pacientes con MM que en controles pareados. Los investigadores suecos también encontraron que el riesgo de infecciones ha aumentado en las últimas décadas, y argumentan que el uso de medidas de tratamiento más intensivas para MM (es decir, medicamentos más nuevos y quimioterapia de dosis alta con trasplante) ha contribuido a un mayor riesgo.
Referencias


")


")